2022 年《化妆品监管现代化法案》MoCRA(Modernization of Cosmetics Regulation Act of 2022)要求在美国境内销售化妆品的制造商(Manufacturers)和加工商(Processors)必须向美国食品药品监督管理局 FDA 注册其生产设施,并每两年更新一次注册信息。责任人必须向 FDA 列名每种上市化妆品产品,包括产品成分,并每年更新相关信息。
化妆品及 FDA 监管权限
美国食品药品监督管理局 FDA 最初仅被授权监管食品和药品(包括兽用食品和药品),但 1938 年《联邦食品、药品和化妆品法案》(Federal Food, Drug, and Cosmetics Act)将化妆品(Cosmetics)、医疗器械(Medical devices)和着色剂(Color additives)纳入了该机构的监管范围。1938 年 6 月 25 日,时任美国总统罗斯福签署了《食品、药品和化妆品法案》,新法律将化妆品和医疗器械纳入监管,并要求药品必须贴上足够详尽的安全使用说明,此外,它还要求所有新药在上市前必须获得批准,制造商必须向 FDA 证明药品是安全的,然后才能销售。FDA 下属的食品安全与应用营养中心 CFSAN(Center for Food Safety and Applied Nutrition)的化妆品和色素办公室(Office of Cosmetics and Colors)负责监管化妆品项目(Ccosmetic program)。

根据《联邦食品、药品和化妆品法案》第 201(i) 条(Section 201(i) of the Federal Food, Drug, and Cosmetic Act)的规定,化妆品(Cosmetics)是指:(1)旨在涂抹、浇淋、喷洒或喷雾于人体或其任何部位,或以其他方式施加于人体或其任何部位,以达到清洁、美化、提升吸引力或改变外观目的的物品;以及(2)旨在用作任何此类物品组成部分的物品,但该术语不包括肥皂(Soap)。
FD&C 法案 201(i)(2) 节将肥皂排除在化妆品的定义之外,符合“肥皂”(Soap)定义的产品不受 FD&C 法案条款的约束。FDA 将“肥皂”一词的适用范围限定于以下情况:
该产品中大部分非挥发性物质由脂肪酸的碱金属盐组成,该产品的洗涤性能归因于碱金属脂肪酸化合物,并且 -
该产品仅作为肥皂进行贴标签、销售和宣传 [CFR 第 21 编第 701.20 部分](21 CFR 701.20)。
符合此类肥皂定义的产品受美国消费品安全委员会 CPSC 的监管,而不受 FDA 的监管。
根据 FD&C Act 法案第 201(i) 条定义涵盖的受 FDA 监管的化妆品包括:纹身贴纸(Tattoos)、脱毛产品(Hair removal products)、润肤霜(Skin moisturizers)、香水(Perfumes)、口红(Lipsticks)、指甲油(Fingernail polishes)、眼部和面部彩妆(Eye and facial makeup)、清洁洗发水(Cleansing shampoos)、沐浴露(Shower gel)、发胶(Hairspray)、烫发剂(Permanent waves)、染发剂(Hair colors)、除臭剂(Deodorants)、部分口腔护理产品(如牙膏,漱口水和口气清新剂)、某些清洁湿巾(Cleansing wipes),以及任何拟用作化妆品成分的物质。
但是,如果产品旨在用于治疗用途,例如治疗或预防疾病,或影响人体结构或功能,则即使其影响外观,也属于药品(Drug)(FD&C Act, 201(g)),某些情况下也可能属于医疗器械(Medical device)(FD&C Act, 201(h))。其他“个人护理产品”(Personal care products)可能被作为膳食补充剂(Dietary supplements)或消费品(Consumer products)进行监管。
FDA 对化妆品的法定监管权限与其他受监管产品(例如药品、生物制品和医疗器械)不同。根据美国联邦法律规定,化妆品及其成分无需获得 FDA 的上市前批准(Premarket approval),但着色剂除外(Color additives)。然而,对于不符合法律规定的市场在售产品,或违反法律的企业及个人,FDA 可采取执法行动。即对于化妆品,FDA 不做上市前批准(着色剂除外),而是事后监管。
一般来说,除着色剂(Color additives)及法规禁止和限制使用的成分(Ingredients)外,制造商可在化妆品配方中使用任何成分,但须满足以下条件:
该成分和成品化妆品在标签所示或常规使用条件下是安全的; 产品标签规范;以及 -
使用该成分不会导致化妆品违反美国食品药品监督管理局 FDA 执行的法律规定,构成掺假或误标。
在美国销售含有着色剂(Color additive)的产品之前,必须确认该着色剂是否获准用于其预期用途(Intended use)。需认证着色剂(Certifiable color additives),有时也称为“合成有机色素”(Synthetic-organic colors),必须经过美国食品药品监督管理局 FDA 的批次检测(Batch testing)。这一流程称为着色剂认证(Color additive certification),旨在确保着色剂在用于化妆品之前,其安全性、质量、一致性和着色强度均符合要求。《联邦法规汇编》第 21 编第 74 部分 C 分部以及第 82 部分 B 和 C 分部(Part 74, Subpart C and Part 82, Subparts B and C)列明了化妆品中允许使用的、需进行批次认证的着色剂。当 FDA 对一批散装着色剂进行认证后,会向该着色剂制造商(Manufacturer)分配一个唯一的六位批号,批号以两个字母开头,后跟四个数字(例如 XX1234)。成品制造商(Finished-product manufacturers)必须确保其从着色剂制造商处采购的着色剂已通过批次认证,并标注有相应的批次认证批号,才能在美国合法用于销售的产品中。
化妆品中添加的每种着色剂均须列入产品成分声明中(可采用全称或缩写名称,例如:Ext. D&C Yellow No. 7 或 Ext. Yellow 7)。FDA 可通过其访问其内部着色剂认证数据库(FDA’s Color Certification database),输入六位数的批号来验证每个着色剂认证批号的真伪。
根据《联邦食品、药品和化妆品法案》第 801(a) 条(Section 801(a) of the FD&C Act)的规定,进口化妆品在通过美国海关入境时须接受 FDA 的审查,不符合 FDA 法律法规的产品将被拒绝入境美国。
FDA 进口拒绝报告 IRR(Import refusals report)按国家/地区和产品(基于行业代码)(Industry code)列出进口拒绝的产品信息列表。IRR 每月更新一次,目的是向公众提供有关疑似违反 FDA 法律法规的货物信息。
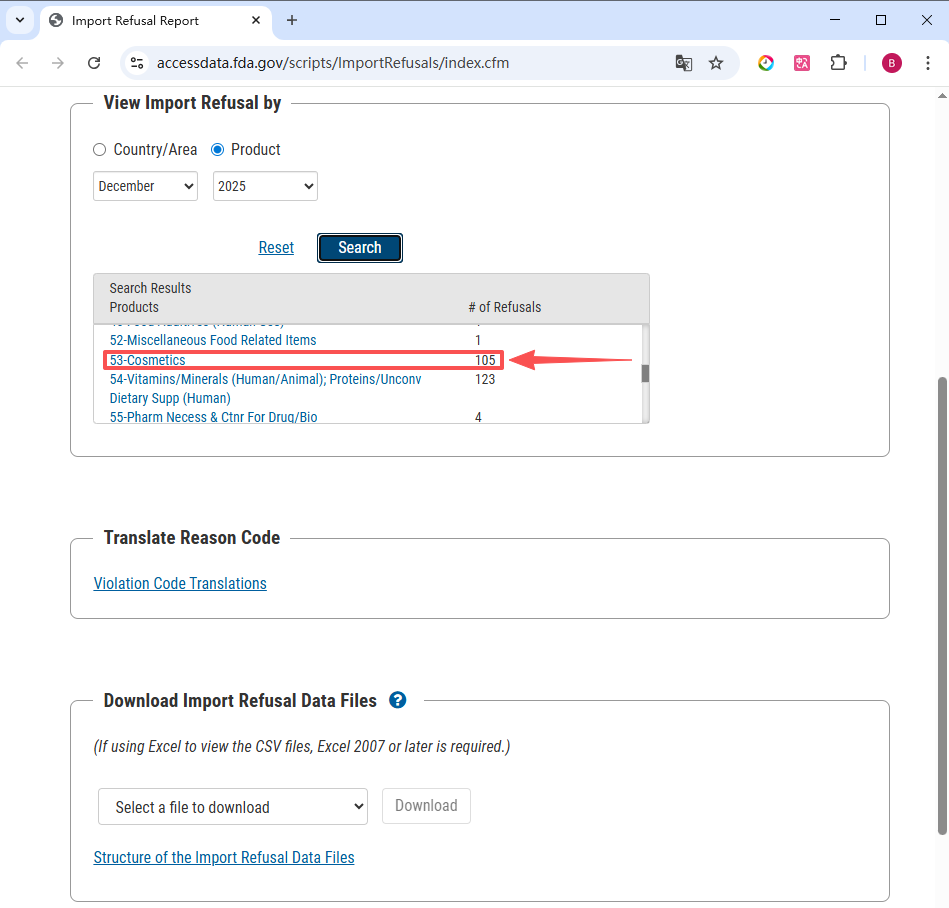
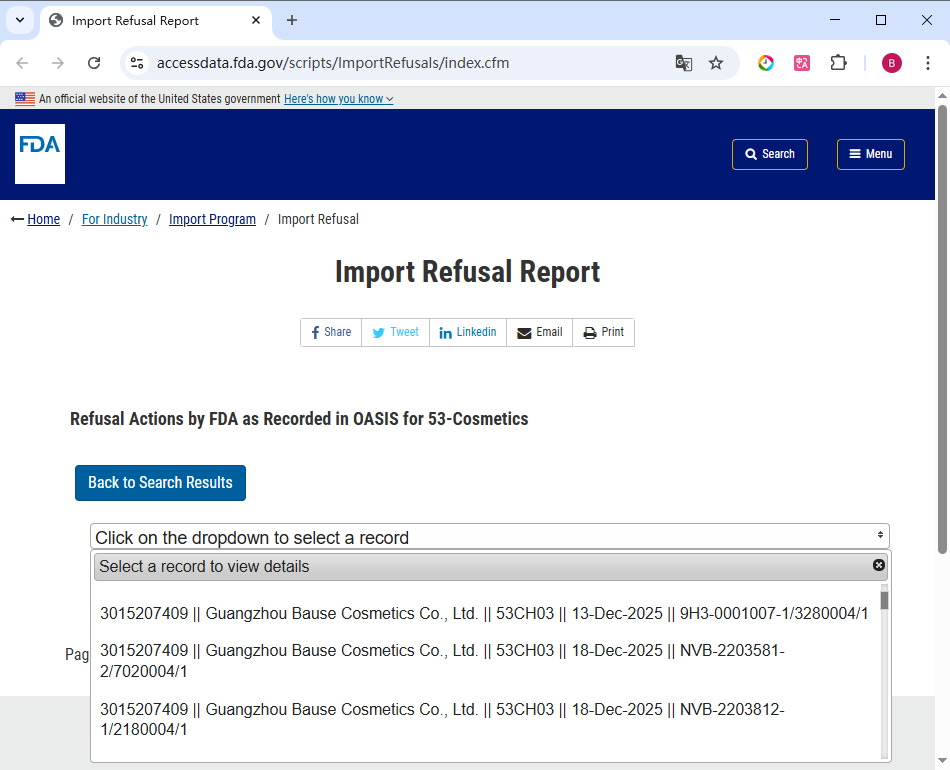

点击便可查看 FDA 化妆品进口拒绝记录的详情信息。
化妆品设施注册和产品列名
在美国,联邦法律由美国国会(Congress)制定。为了使法律在日常运作中有效执行,美国国会授权某些政府机构(例如美国食品药品监督管理局 FDA)制定法规(Regulations)。如果要改变 FDA 对化妆品的法定监管权限,则需要美国国会修改相关法律。
作为 《2023 财年综合拨款法案》(Consolidated Appropriations Act, 2023)(Pub. L. 117-328)的一部分,《2022 化妆品监管现代化法案》MoCRA(Modernization of Cosmetics Regulation Act of 2022)于 2022 年 12 月 29 日正式签署成为法律,这是自 1938 年《联邦食品、药品和化妆品法案》(Federal Food, Drug, and Cosmetic Act)通过以来,美国食品药品监督管理局 FDA 对化妆品监管权限的最大一次扩展。
除其他条款外,MoCRA 在《联邦食品、药品和化妆品法案》中新增了第 607 条(Section 607),规定了化妆品设施注册和产品列名的要求。



-
设施注册(Facility Registration):
制造商(Manufacturers)和加工商(Processors)必须向美国食品药品监督管理局 FDA 注册其设施,并每两年更新一次注册。设施(Facility)是指任何制造或加工在美国境内分销的化妆品的场所,包括进口商所设立、并从事上述制造或加工活动的场所。本质是“受监管的化妆品生产/加工场所”。 -
产品列名(Product Listing):
责任人(Responsible person)必须向 FDA 列名每款上市的化妆品产品,包括产品成分,并每年提供更新信息(如配方变更,成分比例变化,标签重大变化等)。责任人(Responsible person)是指按照《联邦食品、药品和化妆品法案》第 609(a) 条(Section 609(a) of the FD&C Act)或《公平包装与标签法案》第 4(a) 条(Section 4(a) of the Fair Packaging and Labeling Act)的规定,其名称标示在该化妆品标签上的化妆品制造商(Manufacturer)、包装商(Packer)或分销商(Distributor)。
此外,对于外国设施(位于美国境外),注册时还必须指定一名美国代理(U.S. agent),美国代理是指居住在美国或在美国设有营业场所并实际身处美国境内的个人或商业实体。
根据《联邦食品、药品和化妆品法案》第 607 条(Section 607 of the FD&C Act)向 FDA 提交化妆品设施注册或产品列名无需任何费用,可通过 FDA 的 Cosmetics Direct 系统直接免费以电子方式提交。FDA 的经费来源主要是美国国会授权的预算和用户收费项目(比如药品审评费、医疗器械审查费等),但化妆品监管目前不属于此类有用户收费的项目。
FDA 化妆品设施注册需要提交哪些信息
根据《联邦食品、药品和化妆品法案》第 607(a) 条和第 607(b)(2) 条(Sections 607(a) and 607(b)(2) of the FD&C Act)的规定,化妆品设施注册时必须提交以下信息:
设施所有者和/或经营者的名称; 设施名称、实际地址、电子邮件地址和电话号码; 对于任何外国设施,需提供该设施的美国代理的联系方式(姓名和电话号码),以及(如有)电子联系信息(电子邮件); 该设施先前由 FDA 分配的设施注册号(如有); 该设施生产或加工的化妆品销售所使用的所有品牌名称; 该设施生产或加工的每种化妆品的产品类别及责任人; -
提交类型(Type of submission)。
FDA 化妆品产品列名需要提交哪些信息
根据《联邦食品、药品和化妆品法案》第 607(c) 条(Section 607(c) of the FD&C Act)的规定,化妆品产品列名必须提交以下信息:
生产或加工化妆品的每处设施的设施注册号(Facility registration number); 责任人姓名及联系方式,以及化妆品名称(以标签上显示的名称为准); 该化妆品产品适用的化妆品类别; 化妆品成分清单,包括任何香料、香精或色素,每种成分均须按《联邦法规汇编》第 21 编第 701.3 条(Section 701.3 of title 21, Code of Federal Regulations)(或其任何后续法规)的要求,以成分名称或通用名称进行标识; 产品列名编号(如有先前分配);以及 -
提交类型(Type of submission)。
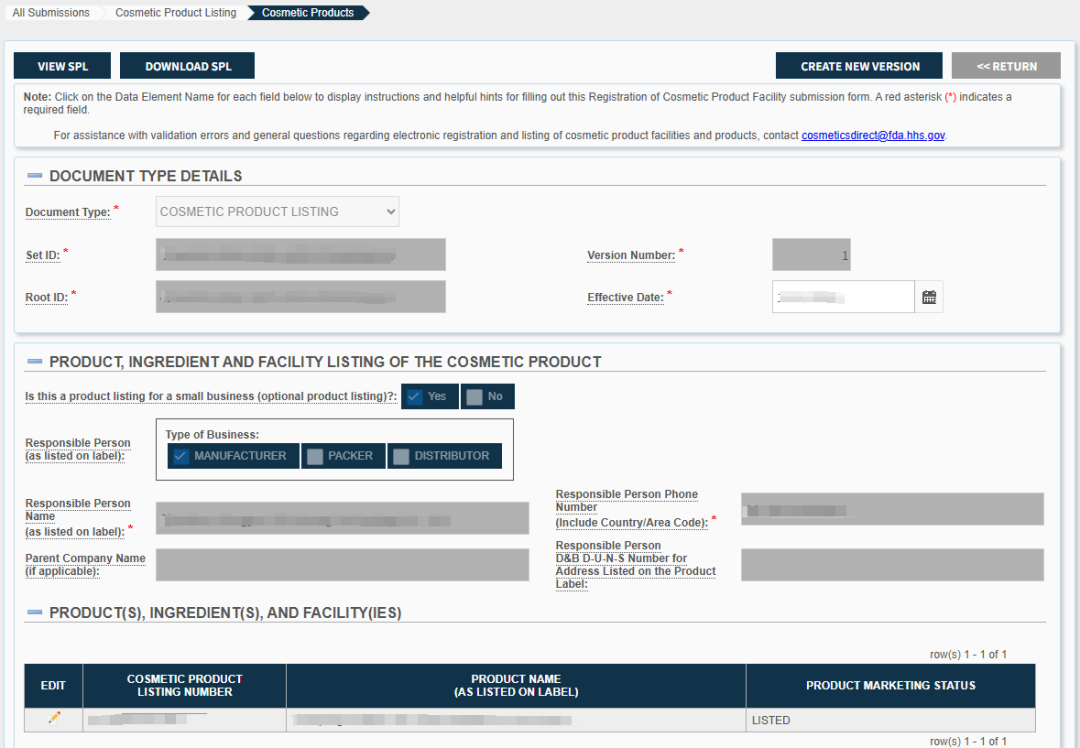
根据《联邦食品、药品和化妆品法案》第 607(c)(4)(B) 条(Section 607(c)(4)(B) of the FD&C Act)的规定,一次化妆品产品列名提交可以涵盖多个化妆品产品,前提是这些产品的配方完全相同,或者仅在颜色、香型或风味方面存在差异,或仅在净含量方面不同。
FDA 还要求提交以下可选的附加信息:
母公司名称(如适用); 业务类型(如标签上所列),即制造商(Manufacturer)、包装商(Packer)或分销商(Distributor); 标签图片(目前仅接受 jpg 格式文件); 产品网页链接; 该化妆品是否仅供专业人士使用; 产品标签上所列地址的责任人 DUNS 编号; 唯一成分标识符 UNIIs(Unique Ingredient Identifiers);以及 -
与该产品列名相关的其他个人联系方式。
化妆品设施注册号
化妆品设施注册号(Facility registration number)即为 FDA 机构识别号 FEI(FDA Establishment Identifier)(7 - 10 位)。FEI 也称为企业或设施机构识别号(Firm or Facility Establishment Identifier),是由 FDA 分配的唯一标识符(Unique identifier),用于识别与 FDA 监管产品相关的企业。
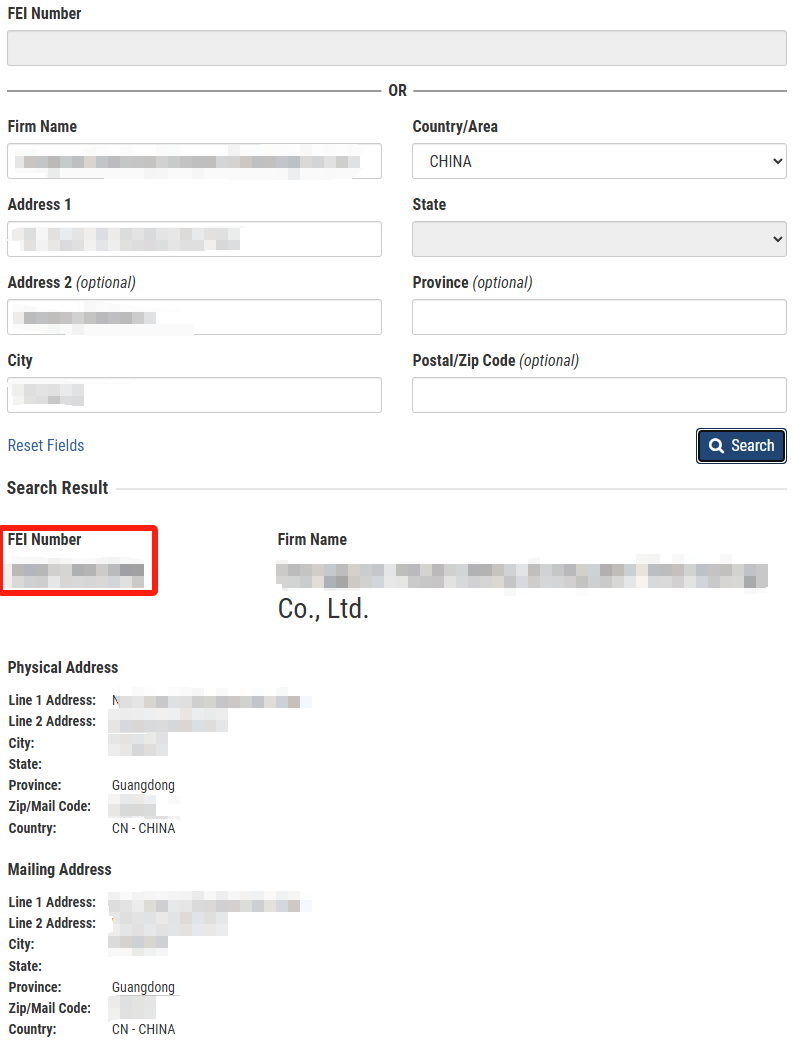
要确定某实体(Entity)是否已拥有 FEI 编号,可通过 FDA 的 FEI 搜索门户(FEI Search Portal)在线搜索查询。
化妆品设施注册&产品列名电子提交
《联邦食品、药品和化妆品法案》第 607(a) 条(Section 607(a) of the FD&C Act)规定,凡拥有或经营从事用于在美国境内分销的化妆品的制造或加工活动的设施之任何主体,均须向美国食品药品监督管理局 FDA 为其每一处该等设施进行注册。该法案第 607(c) 条(Section 607(c) of the FD&C Act)进一步规定,对于每一种化妆品产品,责任人必须向 FDA 进行产品列名。但根据该法案第 612 条(Section 612 of the FD&C Act)所界定的特定小型企业,可豁免上述设施注册及产品列名的要求。
简单来说,即凡是生产或加工销往美国的化妆品的工厂,必须向 FDA 进行设施注册;同时,每一款在美国销售的化妆品产品,必须由责任人向 FDA 进行产品列名;只有极少数符合法定条件的小型企业,才能同时豁免这两项义务。
FDA 已于 2023 年 3 月 27 日终止了自愿性化妆品注册计划 VCRP(Voluntary cosmetic registration program),停止接受和处理化妆品机构(Cosmetics establishments)和产品自愿注册计划(Voluntary registration program)的电子版和纸质提交(Electronic and paper submissions)。同时建立一套新系统,其中包括一个用于提交化妆品生产设施注册和产品列名的门户网站,以满足 MoCRA 法案规定的设施注册(Facility registrations)和产品列名(Product listings)要求。
2023 年 10 月 18 日,美国食品药品监督管理局 FDA 宣布推出 Cosmetics Direct,用于化妆品生产设施和产品的电子注册和列名。除电子提交(Electronic Submissions)外,2024 年 1 月 8 日,FDA 还宣布推出纸质表格 FDA 5066 表(Form FDA 5066:Registration of Cosmetic Product Facility)和 FDA 5067 表(Form FDA 5067:Cosmetic Product Listing)作为向 FDA 提交化妆品生产设施注册和化妆品产品列名的替代提交工具。
Cosmetics Direct 是用于 FDA 化妆品生产设施注册和产品列名的电子提交门户网站(Electronic submissions portal),旨在协助简化根据《联邦食品、药品和化妆品法案》第 607 条(Section 607 of the FD&C Act)提交和接收注册及产品列名信息的流程。它是 FDA 提供的一款结构化产品标签 SPL(Structured Product Labeling)撰写工具,用于化妆品生产设施注册和化妆品产品列名。该工具包含用户友好的数据输入表单(Data entry forms),可执行初始验证,创建并保存 SPL 提交内容,并直接向 FDA 提交 SPL 进行内部处理,无需使用电子提交网关 ESG(Electronic Submissions Gateway)。结构化产品标签 SPL(Structured Product Labeling)是一种经 HL7(Health Level Seven)批准,并被美国食品药品监督管理局 FDA 采纳的文档标记标准(Document markup standard),用于在监管系统中交换产品及生产设施相关信息。

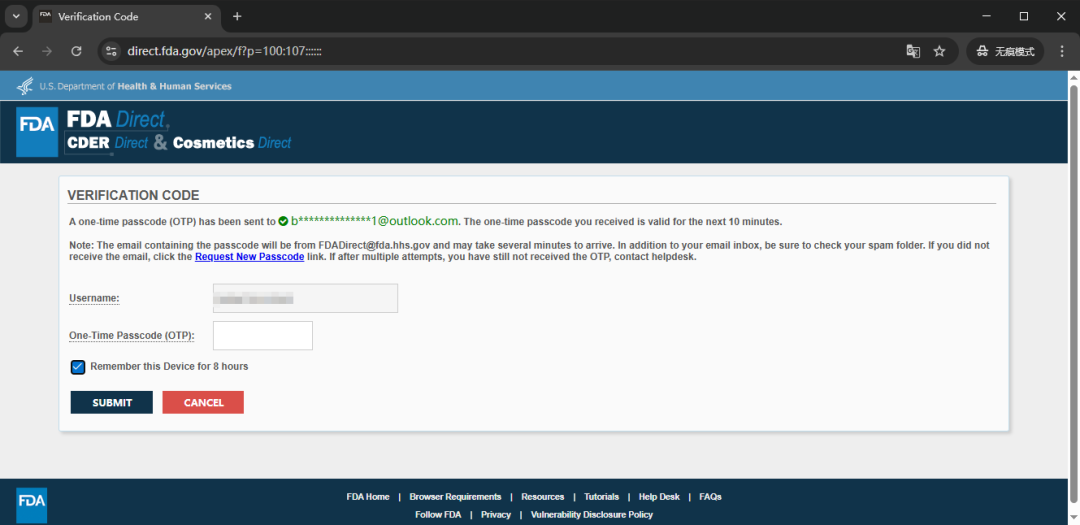
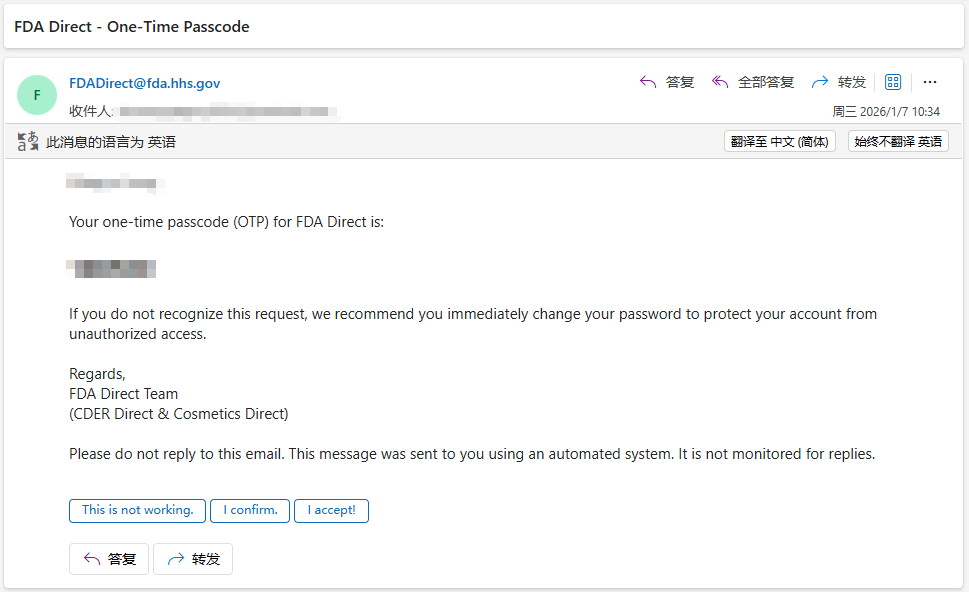
查看账户注册的电子邮件,获取一次性密码 OTP(One-Time Passcod),然后在页面的 OTP 字段的输入框中输入,
同时勾选 OTP 输入框下的“Remember this Device for 8 hours”(记住此设备 8 小时)复选框,勾选此复选框将阻止在 8 小时内出现验证步骤,如果不勾选此复选框,则每次登录 FDA Direct 时都必须重新完成上述 OTP 验证码验证步骤,
最后点击 ”Submit“ 提交即可登录 FDA Direct,所有帐户均有 30 分钟的会话超时限制,如果超过 30 分钟未进行任何操作,系统将自动从 FDA Direct 登出,
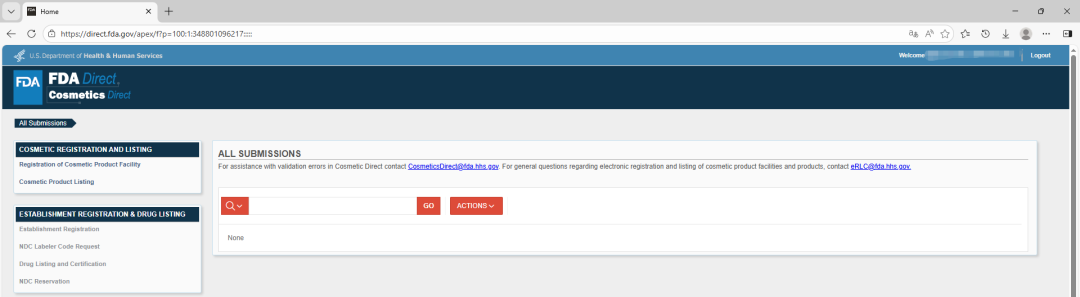
Cosmetics Direct 账户登录后,后台仪表盘左侧菜单显示 FDA Direct 中所有可用的提交表格。某些表格的访问权限受限,具体取决于你的账户类型以及你在创建账户流程中取消选择的任何选项。菜单中灰色区域表示你无权访问特定表格或表格组。
如果你此前从未提交过机构注册(Establishment registration)、产品列名(Product listing)等,则可以通过 FDA Direct 中的标准 SPL 提交模板创建新的提交(Submission)。FDA Direct 提供多种空白模板,适用于不同类型的提交。
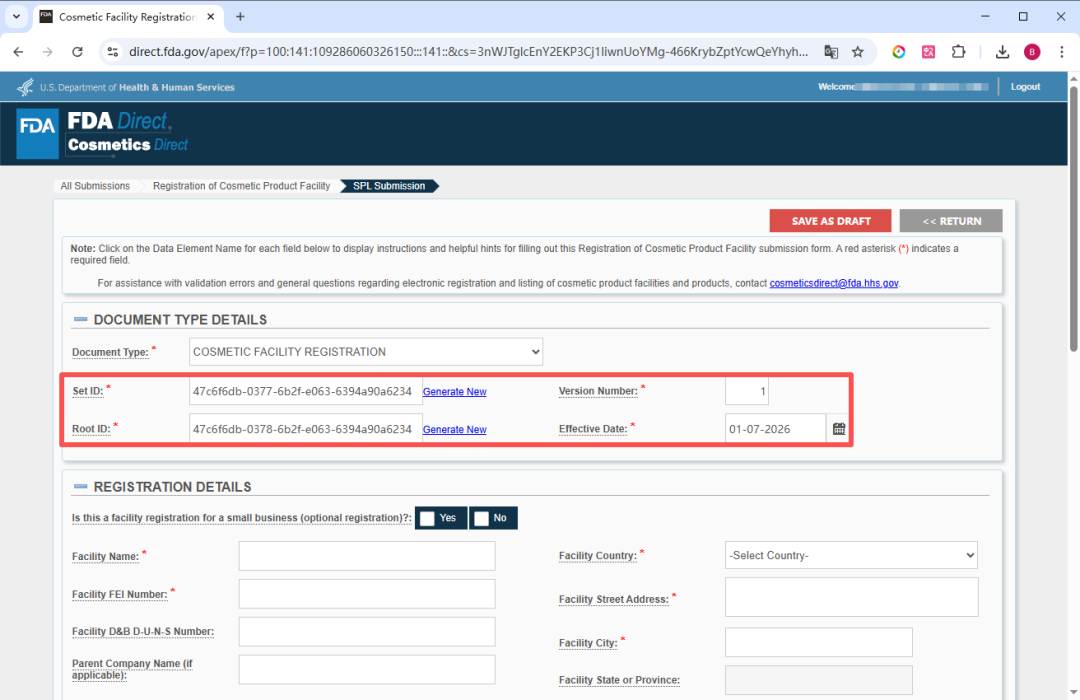
创建新提交时,每份提交文件的顶部都包含一组预先生成的信息,即提交标头信息(Submission Header Information):
-
Set ID:
此字段(Field)由系统自动生成。Set ID 用于唯一标识 SPL 提交中的一组版本,对于每个提交“集”(Set)(即一组提交版本)都保持不变,当你提交同一提交(Submission)的不同版本时,Set ID 在每个新版本中都保持不变。当 SPL 提交发生更改时,系统会为新的 SPL 提交分配一个新的 Root ID,但同时也会使用原始 SPL 提交中的 Set ID。Set ID 是一个全局唯一标识符 GUID(Globally Unique Identifier),GUID 是由数字和小写字母组成的字符串,使用特定定义的数学算法(Specifically defined mathematical algorithm)生成,以确保在同一系统中使用相同 GUID 的概率极低。例如:9aa9d2e6-6982-48e5-831d-dbe7c04a14ed。 -
Root ID:
此字段(Field)由系统自动生成。Root ID 用于唯一标识特定的 SPL 文件(SPL file)。当你创建新提交或提交先前提交的新版本时,Root ID 每次都会改变(与 Set ID 不同),即每个 SPL 文件的新版本都会生成一个新的 Root ID。Root ID 是一个全局唯一标识符 GUID(Globally Unique Identifier),GUID 是由数字和小写字母组成的字符串,使用特定定义的数学算法(Specifically defined mathematical algorithm)生成,以确保在同一系统中使用相同 GUID 的概率极低。例如:9aa9d2e6-6982-48e5-831d-dbe7c04a14ed。 -
Version Number:
版本号用于对 SPL 提交的不同版本提供顺序编号。版本号是大于零的整数,例如 6、7 或 8。每次对 SPL 提交进行更改时,版本号都会递增。“版本号”字段中需输入大于 0 的数字。 -
Effective Date:
生效日期。即本表格的创建日期,用户可以修改提交文件的创建日期。但是,系统仅采用实际提交给 FDA 的注册日期。SPL 提交后,用户将无法编辑此日期。

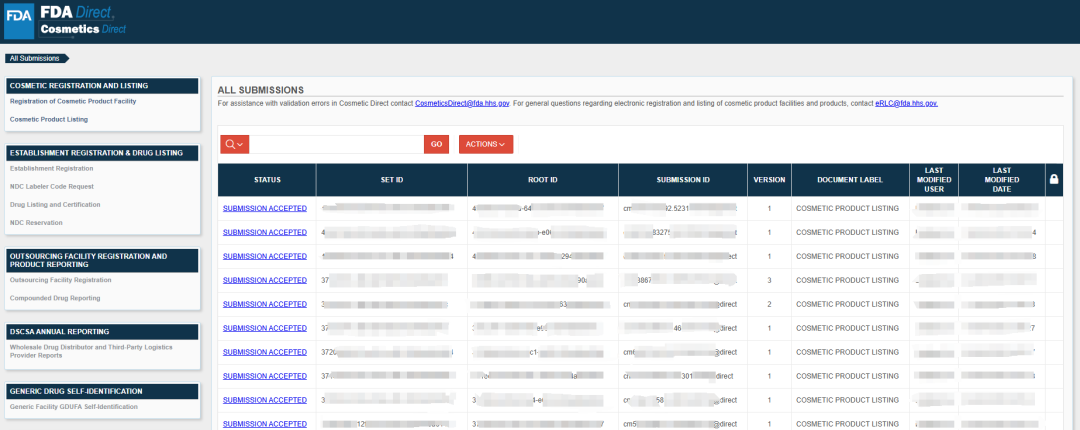
如果你的 SPL 提交(SPL submission)已成功提交至 FDA,则其状态栏中将显示“提交已接受”(Submission accepted)状态。至此,Cosmetics Direct 提交流程已完成,除非你需要对提交信息进行任何更改,否则无需采取任何进一步行动。
最后,美国食品药品监督管理局 FDA 不会为化妆品生产设施注册或化妆品产品列名颁发“证书”(Certificates)。化妆品机构(Establishment)的注册、机构注册号的分配或化妆品产品的列名,并不意味着 FDA 已批准该企业或其产品(参见 21 CFR 710.8 和 720.9),也不意味着该产品符合《联邦食品、药品和化妆品法案》(FD&C Act)中对化妆品(Cosmetic)的定义。任何因注册或持有注册号而在标签或广告中暗示官方批准(Official approval)的行为均被 FDA 视为误导性宣传。
以上便是关于 FDA 监管产品化妆品及化妆品设施注册和产品列名的介绍。
文章为作者独立观点,不代表DLZ123立场。如有侵权,请联系我们。( 版权为作者所有,如需转载,请联系作者 )

网站运营至今,离不开小伙伴们的支持。 为了给小伙伴们提供一个互相交流的平台和资源的对接,特地开通了独立站交流群。
群里有不少运营大神,不时会分享一些运营技巧,更有一些资源收藏爱好者不时分享一些优质的学习资料。
现在可以扫码进群,备注【加群】。 ( 群完全免费,不广告不卖课!)







发表评论 取消回复