510(k) 是向 FDA 提交的上市前申报(Premarket submission),旨在证明拟上市的新型医疗器械(New device)与已合法上市且无需 PMA 批准的医疗器械具有同等的安全性和有效性,即实质等同 SE(Substantially equivalent)。除非该医疗器械符合 510(k) 豁免条件,否则必须在上市前至少 90 天向 FDA 提交 510(k)。510(k) 法规(510(k) regulation)载于《联邦法规汇编》第 21 编第 807 部分 E 节(21 CFR 807 Subpart E),其中规定了 510(k) 申报所需提交的信息。所有医疗器械的 510(k) 申报均采用基本相同的格式,并包含相同的基本信息(必备要素),但 510(k) 申报并非固定表格(The 510(k) is not a form),相关信息应足够详细,应以条理清晰、表格化的文档形式提交,以便 FDA 能够确定该医疗器械与已合法上市(Legally marketed)的类似医疗器械实质等同 SE(Substantially equivalent),部分章节仅需一页篇幅,其他章节可能达 50 页以上,FDA 表示,平均每份 510(k) 申请约 35 页,复杂医疗器械的申请可能超过 100 页,具体取决于医疗器械的复杂程度。
FDA 医疗器械分类
美国食品药品监督管理局 FDA 根据风险程度对医疗器械进行监管。
1976 年 5 月 28 日,美国《联邦食品、药品和化妆品法案》(FD&C)(Federal Food, Drug, and Cosmetic Act)《医疗器械修正案》(MDA)(Medical Device Amendments)(Pub. L. 94-295)颁布。MDA 源于美国参议院(U.S. Senate)的一项调查结果,该调查认定有缺陷的医疗器械(Faulty medical devices)已造成 10000 起伤害事件,其中包括 731 例死亡。
MDA 要求美国食品药品监督管理局 FDA 制定法规,根据为合理确保(Reasonable assurance)其安全性和有效性所需的监管程度,将当时处于商业流通(Commercial distribution)的所有医疗器械分为三个监管控制类别之一:I 类、II 类或 III 类。医疗器械所属类别决定了医疗器械制造商在州际贸易(Interstate commerce)中分销该器械之前必须满足的合规要求。
根据 FD&C 法案第 513(a)(1) 条(Section 513(a)(1) of the FD&C Act)(21 U.S.C. § 360c(a)(1)),这三个医疗器械类别定义如下:
-
I 类(Class I):
被视为低风险医疗器械(Low risk devices),如弹性绷带(Elastic bandages),约 47% 的医疗器械属于此类,须遵守一套适用于所有类别医疗器械的全面监管要求,称为一般管制要求(General controls)。大多数 I 类医疗器械(但并非所有) 可免于上市前通知/510(k) 要求。 -
II 类(Class II):
被视为中等风险医疗器械(Moderate risk devices),如电动轮椅(Powered wheelchairs),约 43% 的医疗器械属于此类,受一般管制要求和特殊管制要求约束(Subject to general and special controls)。即对于仅凭一般管制要求不足以合理保证其安全性和有效性,且已有充分信息可据此建立特殊管制要求(Special controls)以提供此类保证。大多数(但并非所有)II 类器械在上市前需要获得上市前通知/510(k) 许可(510(k) clearance),部分 II 类医疗器械(Class II devices)可获得 510(k) 豁免(510(k) exempt),只要符合 FDA 规定的特殊管制要求(Special controls)。 -
III 类(Class III):
被视为高风险医疗器械(High risk devices),如可植入式心脏起搏器(Implantable pacemakers),约 10% 的医疗器械属于此类,这类医疗器械通常用于维持或支持(Sustain or support)生命,属于植入类器械,或存在导致疾病或损伤的不合理风险,对于仅凭一般管制要求不足以合理保证其安全性和有效性,且现有信息不足以建立特殊管制要求(Special controls)以提供此类保证,受一般管制要求约束以及在上市前需要获得 PMA 申请的批准。
上市前通知(Premarket notification)是指将新医疗器械(New device)即修正案后器械(Post-amendments device)归入上述三类医疗器械之一的过程。制造商若计划在美国市场销售无需提交上市前批准申请 PMA 供人类使用的 I、II 或 III 类医疗器械,则必须向 FDA 提交上市前通知(Premarket notification)(通常称为 510(k)),除非该医疗器械符合美国《联邦食品、药品和化妆品法案》(FD&C Act)的 510(k) 豁免要求,且未超出每项器械分类法规(21 CFR第862至892部分第.9节)规定的豁免限制(21 CFR 第 862 至 892 部分第 9 节,例如 21 CFR 862.9、21 CFR 864.9 等)(Section .9 of 21 CFR Parts 862 through 892, e.g., 21 CFR 862.9, 21 CFR 864.9, etc.)。根据《联邦食品、药品和化妆品法案》第 510(k) 条(Section 510(k) of the FD&C Act)规定,制造商(Manufacturer)必须在将医疗器械投入州际商业流通进行商业分销之前至少 90 天向 FDA 提交 510(k),以便 FDA 判定该医疗器械是否符合上市许可标准(Criteria for market clearance)(Sections 510(k) and (n) of the FD&C Act (21 U.S.C. §§ 360(k) & (n)))。
FDA 的决定依据是该医疗器械是否与合法上市的对比器械(Predicate)实质等同 SE(Substantially equivalent)(Section 513(i) of the FD&C Act (21 U.S.C. § 360c(i)))。在 FDA 发布命令(510(k) 许可)(510(k) clearance)表明该医疗器械已被确定为 SE 之前(Section 513(f)(1) of the FD&C Act (21 U.S.C. § 360c(f)(1))),该医疗器械不得投入商业流通。
若无法确定医疗器械适用的医疗器械分类(Device classification),可考虑 FDA 513(g) 医疗器械分类咨询项目(513(g) program)。513(g) Program 是向 FDA 提交的信息请求(Request for information),根据《联邦食品、药品和化妆品法案》(FD&C法案)第 513(g) 条(Section 513(g) of the Federal Food, Drug and Cosmetic Act, or the FD&C Act)的规定,FDA 将提供他们认为适用于你的医疗器械的监管途径(Regulatory pathway)作为回应。513(g) 需要支付用户费用(User fee),FDA 对 513(g) 的回应不构成 FDA 对产品的许可或批准(Clearance or approval)。如果你提交了 513(g) 申请,并且 FDA 给出了合适的监管途径,则你仍然必须遵循该途径(Pathway)的适用监管要求,例如提交 510(k) 或提交上市前批准申请(Premarket approval application)。
什么是 510(k)
510(k),也称为上市前通知 PMN(Premarket notification),是医疗器械上市的主要途径之一。实际上,510(k) 是医疗器械最常见的上市前申报方式(Premarket submission)。
之所以将上市前通知称为 510(k),是因为《联邦食品、药品和化妆品法案》第 510(k) 条(Section 510(k) of the Federal Food, Drug, and Cosmetic Act)规定,必须注册的医疗器械制造商应至少提前 90 天向 FDA 提交其上市医疗器械的意向通知,即上市前通知(Premarket notification)PMN 或 510(k)。该程序使 FDA 能够判定该医疗器械是否与已归入三个分类类别之一的医疗器械实质等同。因此,尚未分类的新型医疗器械(New device)(1976 年 5 月 28 日之前未上市销售)可以得到正确识别和分类。
510(k) 被视为上市前许可申请(Marketing clearance application),由 FDA 负责审批,该许可(Clearance)基于“实质等同”(Substantial Equivalence)的判定,需要说明的是,510(k) 不仅仅是一份表格,它可能包含数百页的完整申报材料,具体取决于医疗器械类型及证明实质等同所需提交的内容。
510(k) 申请获得的是 FDA 核准(clearance),即获得 FDA 510(k) 上市许可(FDA cleared),该医疗器械已通过 FDA 510(k) 审查,允许在美国市场销售。510(k) 不是机构注册或医疗器械列名(Establishment registration or device listing),也不是上市前批准 PMA(Premarket approval)。因此,宣称 FDA 已核准(Cleared)你的 510(k) 申请,进而认定产品获得 FDA 批准(DA approved),这种说法并不恰当。唯有通过上市前批准程序(Premarket approval process)获批的(Approved)的医疗器械,才可视为获得 FDA 批准(FDA approved)。
实质等同 SE(Substantial Equivalence)是 510(k) 计划(510(k) program)的核心内容,其本质上是证明:相较于对比器械(Predicate device),你的新型医疗器械具有相同的预期用途(Intended use)和技术特性(Technological characteristics);或具有相同的预期用途,且技术特性差异不会引发关于安全性和有效性的不同问题。证明实质等同的最常用方法是提交传统 510(k)(Traditional 510(k)),由 FDA 进行审核并获得 FDA 的许可(FDA review and clearance)。根据 21 CFR 807.92(a)(3),用于与新型医疗器械进行实质等同比较的已合法上市(Legally marketed)器械,是指 1976 年 5 月 28 日前已合法上市的器械,或经由 III 类重新分类为 II 类或 I 类的器械,或已通过 510(k) 上市前通知流程((510(k) premarket notification process)被认定为实质等同的器械。
如果提交的医疗器械在设计、材料、化学成分、能源、制造工艺或预期用途方面与 1976 年 5 月 28 日之前上市的医疗器械(对比器械)(Predicate device)存在显著差异,则该医疗器械通常需要获得上市前批准 PMA。
510(k) 审查标准(新型器械与已合法上市对比器械实质等同)与 PMA 审查标准(安全性和有效性的合理保证)不同。PMA 审查标准依赖于对安全性和有效性的独立论证,而 510(k) 审查的法定标准(Statutory standard)是比较性审查,即对新型医疗器械与对比器械进行比较,证明拟上市的医疗器械与已合法上市的对比器械(Predicate device)实质等同 SE(Substantial equivalence),经认定实质等同的 510(k) 申报医疗器械,即可在美国合法上市销售。尽管如此,安全性和有效性原则仍是每项 510(k) 审查中实质等同判定的基础,510(k) 审查中实质等同的判定标准载于《联邦食品、药品和化妆品法案》第 513(i) 条(Section 513(i) of the FD&C Act)。
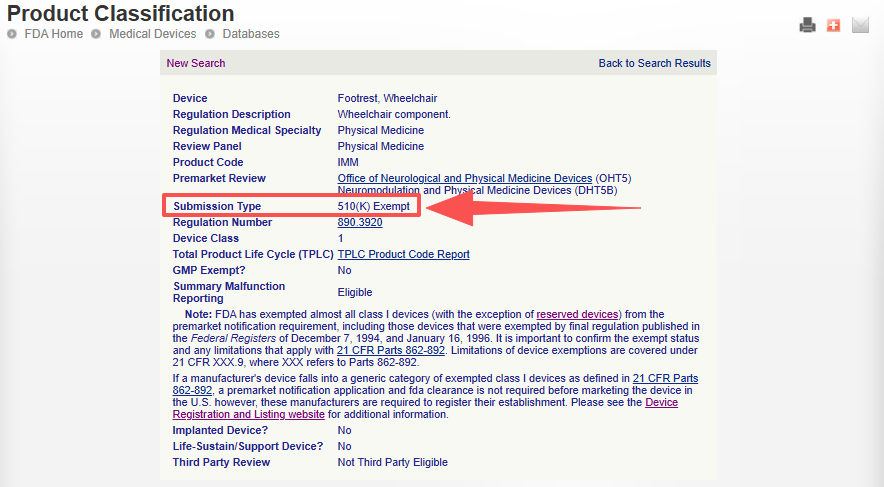
此外,对于获得 510(k) 豁免的 I 类器械,无需提交 510(k) 申报(Submission of a 510(k))和获得 FDA 上市许可(Marketing clearance from FDA)。
什么是 510(k) 摘要
无论 I 类、II 类还是 III 类医疗器械,所有 510(k) 均需提交摘要或声明(Summary or Statement)。

510(k) 摘要(The 510(k) Summary)是一份文件,即为支持你实质等同主张所依据的信息的概括性陈述,由制造商编制并包含在提交的 510(k) 申报中,必须包含 21 CFR 807.92 中确定的所有要素。510(k) 摘要必须足够详细,以阐明实质等同 SE 认定的依据(21 CFR 807.92(a))。FDA 将在 510(k) 审查期间验证 510(k) 摘要信息的准确性和完整性。
而 510(k) 声明(510(k) Statement)则是 510(k) 申报方(Applicant)作出的书面承诺:在收到书面请求(Written request)后 30 天内,将向任何提出要求的人士提供支持 FDA 实质等同认定所需的安全性与有效性信息。
你的 510(k) 申报必须包含 510(k) 摘要或 510(k) 声明(510(k) Summary or 510(k) Statement),FDA 才能开始对其进行科学审查(Scientific review)。
什么情况下需要 510(k)
通常以下 3 种情况下需要 510(k):第一种情况是首次将新型医疗器械(New device)推向市场时;第二种情况是更改先前已获准医疗器械(Previously cleared device)的适应症(Indications for use);第三种情况是对先前已获批准医疗器械进行重大修改(Making significant modifications)。在这些情况下,就需要提交 510(k),且必须获得 510(k) 许可(510(k) clearance)后产品才能上市销售。
510(k) 上市前申报(510(k) submissions)有 3 种类型:传统 510(k)(Traditional 510(k))、简化 510(k)(Abbreviated 510(k))和特殊 510(k)(Special 510(k))。
尽管基本内容要求适用于所有 510(k) 申报,但证明实质等同所需的数据和信息类型会因医疗器械类型以及新型器械与对比器械(Predicate device)之间的差异而有所不同。FDA 已发布许多针对特定器械的指南文件,阐明特定类型器械的 510(k) 申报中应包含的数据内容。
传统 510(k) 必须提交 21CFR 807.87 规定的必要要素,通常包括企业名称(Business name)、商品名称(Trade name)等 510(k) 申报行政与程序性要求(Administrative requirements)内容。传统 510(k) 适用于任何情况,依赖于实质等同(Substantial Equivalence)的证明,而简化 510(k) 和特殊 510(k) 仅在满足特定条件时方可适用。
简化 510(k) 必须包含 21 CFR 807.87 所规定的必要要素。但简化 510(k) 申报需依据指南文件(Guidance documents)、特殊管制要求(Special controls)及公认共识标准(Recognized standards)。在某些情况下(Under certain conditions),若 510(k) 申报方(Sponsors)遵循 FDA 建议、指南文件或公认共识标准,则无需在简化 510(k) 中提交测试数据(Test data)。在这种情况下,简化 510(k) 申报可能是合适的。
特殊 510(k) 必须包含 21 CFR 807.87 所规定的必要要素。但是,如果你对制造商自有的合法上市医疗器械(Manufacturer's own legally marketed device)进行修改(Modifications)时,则特殊 510(k) 是适用的。这意味着你正在对自有产品进行修改,你拥有该对比器械(Predicate device)的所有权。需特别注意的是,特殊 510(k) 的适用性还要求:修改不得影响医疗器械的预期用途或基本科学技术。
FDA 建议在提交 510(k) 时遵循以下格式提示(Format tips):
由于提交 510(k) 文件可能涉及多方,例如制造商(Manufacturer)、顾问(Consultant)、代理(Correspondent)等,FDA 建议在每次提交时明确说明 510(k) 申报方(510(k) owner)。510(k) 申报方是指对该 510(k) 申报具有法定权限的个人或公司(Individual or firm)。每份 510(k) 仅可由一家公司(Firm)持有。请明确注明 FDA 在审查过程中应联系的负责人,并清晰提供 510(k) 联系人的电子邮箱。在最终决定做出之前,你可以通过电子邮件将联系信息的任何变更通知负责你的 510(k) 的主审员(Lead reviewer)。所有涉及 510(k) 的往来函件均须注明你的 510(k) 编号。 请提供目录(Table of Contents),清晰标明 510(k) 表格中各章节的标题及其对应的页码。目录应包含相应的附件和附录列表。每章另起一页,并用制表符分隔。 为 510(k) 提交中用到的任何图表(Diagrams)、图纸(Drawings)、图示(Figures)、插图(Illustrations)、照片(Photos)、图表(Charts)或表格(Tables)提供标题和/或编号。确保正文中的引用正确指向这些内容。 -
在 510(k) 提交附函(Cover letter)上签名并注明日期。
将所有与 FDA 的通信记录与你的 510(k) 提交副本一起保存,包括 FDA 要求补充信息(如有)、根据 FDA 数据请求提交的任何补充信息,以及你的实质等同证明函件(510(k) 批准函)(510(k) clearance letter)(表示 FDA 已确认实质等同并允许产品上市)的副本。你可能需要这些信息在 FDA 检查员(FDA investigator)检查你的机构(Establishment)时出示。
为便于 FDA 审查申报中的数据、分析及结论,制造商(Manufacturer)应核查以下内容:
数据的逻辑呈现; 测试和数据分析的科学合理性; 测试方案与医疗器械和预期用途的相关性;以及 -
测试或研究摘要报告(Summary report)的完整性。
测试描述及其结果至关重要。所有测试程序(Test procedures)和结果的合理且充分的详细信息均应提交给 FDA。
审查(Review)上市前通知(Premarket notification)后,FDA 将:
发布命令,宣布该医疗器械与合法上市(Legally marketed)的对比器械(Predicate device)实质等同; 发布命令,宣布该医疗器械不与任何已合法上市的对比器械实质等同; 要求提供更多信息;或 暂缓做出决定(Withhold the decision),直至向 FDA 提交证明或披露声明(Certification or disclosure statement); -
告知申报方无需提交上市前通知(Premarket notification),在申报方(Applicant)收到宣布该器械实质等同的命令之前,不得将该器械上市销售。
510(k) 实质等同决定 SE 或非实质等同决定 NSE
FDA 对你提交的 510(k) 申报进行审查后,你可能收到实质等同决定或非实质等同决定。如果 510(k) 申报被认定为实质等同,即表示已获得 FDA 的 510(k) 上市许可(510(k) clearance),该医疗器械可以在美国合法上市。
医疗器械获得 FDA 的上市许可后(Cleared),以下内容/信息将发布在 FDA 的公共 510(k) 数据库中:
实质等同函件(SE Letter) 适应症表(Indications for Use Form) -
510(k) 摘要(510(k) Summary)(若替代 510(k) 声明提交)
如果收到非实质等同决定 NSE(Non-Substantially Equivalent Decision),则可能需要重新提交包含新数据的 510(k)。
常见的一些可能导致 FDA 做出 NSE 决定(NSE Decision)的原因包括:
缺乏对比器械(Predicate device); 医疗器械有新的预期用途(Intended use); 你的医疗器械跟对比器械相比具有不同的技术特性(Technological characteristics),并引发了有关安全性和有效性的不同问题; -
你未能证明你的医疗器械至少与对比器械具有同等的安全性和有效性。
510(k) 申报 FDA 审核简化时间表
日期为日历日;时间表基于 MDUFA III 绩效目标(MDUFA III Performance Goals)。
第 1 天(Day 1):FDA 收到 510(k) 申报(510(k) submission); 到第 7 天(By Day 7):FDA 发送确认函(Acknowledgement Letter)。或者,如果用户费用(User Fee)和/或 eCopy 存在未决问题,FDA 将发送暂缓函(Hold Letter); 到第 15 天(By Day 15):FDA 进行受理性审查(Acceptance Review);FDA 会通知申办方其 510(k) 提交是否被接受进行实质性审查(Substantive Review)或被置于 RTA 暂缓状态(RTA Hold); 到第 60 天(By Day 60):FDA 进行实质性审查(Substantive Review);FDA 通过实质性互动(Substantive Interaction)进行沟通,告知申办方 FDA 将继续进行互动审查流程(Interactive Review);或,将 510(k) 申报置于暂缓状态(Placed on hold)要求提供补充信息(Additional Information); 到第 90 天(By Day 90):FDA 就 510(k) 申报发布最终 MDUFA 决定(MDUFA Decision); -
到第 100 天(By Day 100):如果到第 100 天仍没做出 MDUFA 决定,FDA 将发出《未达成 MDUFA 决定通知》(Missed MDUFA Decision),其中列明待解决的审核问题(Review issues)。
510(k) 用户费用
美国联邦法律授权 FDA 对医疗器械产品审查收取费用,这些费用适用于上市前通知 510(k)、上市前批准申请 PMA 以及产品开发协议 PDP 等,自 2002 年以来,CDRH 开始实施医疗器械用户付费计划(Medical device user fee program),
在用户费用(User fee)制度下,医疗器械企业在向美国食品药品监督管理局 FDA 提交上市前申报(Premarket submission)以在美国销售新医疗器械(New medical device)时,以及向 FDA 进行机构注册和器械列名时,均需缴纳费用,
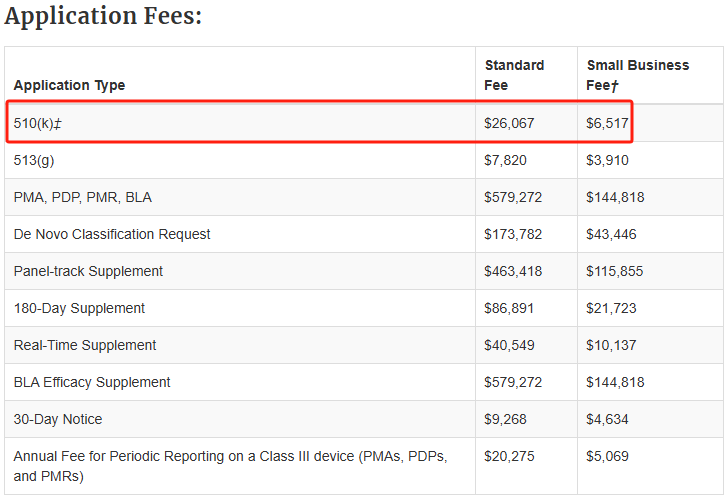
510(k) 申报(510(k) Submissions)用户费用(User Fees)必须在申请提交之日或之前完成缴纳,否则该 510(k) 申报 FDA 将不予受理。
以上便是关于 FDA 医疗器械什么是上市前通知 510(k) 及 510(k) 摘要的介绍。
文章为作者独立观点,不代表DLZ123立场。如有侵权,请联系我们。( 版权为作者所有,如需转载,请联系作者 )

网站运营至今,离不开小伙伴们的支持。 为了给小伙伴们提供一个互相交流的平台和资源的对接,特地开通了独立站交流群。
群里有不少运营大神,不时会分享一些运营技巧,更有一些资源收藏爱好者不时分享一些优质的学习资料。
现在可以扫码进群,备注【加群】。 ( 群完全免费,不广告不卖课!)







发表评论 取消回复